Methods and presentation details
Data sources
The EST contigs are from the
GeneNest assembly
[2]
of the Mar 2001 version of the NCBI
UniGene
[5]
clustering of human genes.
The chromosomes are the Apr 1, 2001 freeze of the
HUGO Golden Path assembly
[6]
of the complete human genome.
Mapping and alignment
The matching pairs of EST contigs and chromosome fragments were
found by searching all matches of length 100 with at most 3 errors
(mismatches or indels) of all contigs of all clusters against the complete genome.
This was done using the fast search program/algorithm vmatch by
Stefan Kurtz.
[7]
The algorithm exploits a modified suffix tree data structure.
Before searching, repeat elements were filtered out using the program RepeatMasker by A.F.A. Smit and P. Green.
For each mathing cluster a refined search against the matching chromosome was made, in order to approximately determine a gene region containing all exons. In the refined search, all matches of length 30 with at most 2 errors were found.
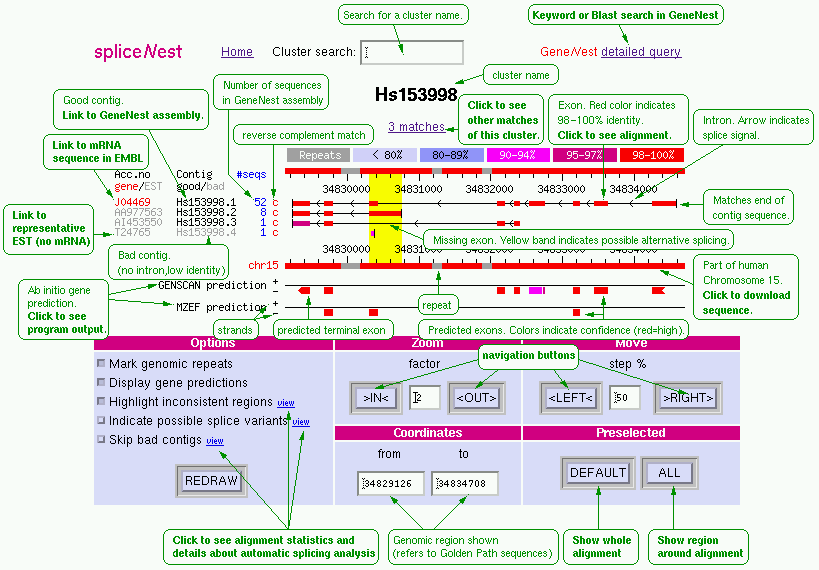
Finally, for each cluster a spliced alignment of all contigs against the matching chromosome region(s) was determined using the program sim4 by Florea et al. [8] The exon positions, percentage identity and splice signals from this aligment are shown in the graphics.
The main criterion for including a match is that it contains a 100 bp region with at most 3 errors. Most such matches are aligned, but the following exceptional cases are skipped:
When a cluster has more than one match, a table of all matches is shown when the cluster is searched. (When browsing matches, use the link containing the number of matches, just above the graphics, to display this table.) The matches are sorted by quality, starting with the best, according to the follwing criteria (by priority):
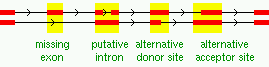
When browsing matches in a chromosome, it is possible to select only matches where alternative splice candidates are found (that is, there are yellow bands).
Only the good contigs are considered when detecting splice variants. If a match contains at least two good contigs, overlapping by at least 100 bp in the genomic sequence, the splice sites according to the alignments are compared and inconsistencies reported as possible splice variants. They are classified as missing exons, putative introns, or alternative donor/acceptor sites (must differ by at least 9 bp to be marked), or as combinations of these.
| [1] | Coward,E., Haas,S.A., and Vingron,M. (2002). SpliceNest: visualization of gene structure and alternative splicing based on EST clusters. Trends Genet., 18 (1), 53-55. |
| [2] | Haas,S.A., Beissbarth,T., Rivals,E., Krause,A., and Vingron,M. (2000). GeneNest: automated generation and visualization of gene indices. Trends Genet. 16 (11), 521-523. |
| [3] | Krause,A., Stoye,J., and Vingron,M. (2000) The SYSTERS Protein Sequence Cluster Set. Nucleic Acids Res. 28 (1), 270-272. |
| [4] | Krause,A., Haas,S.A., Coward,E., and Vingron,M. (2002). SYSTERS, GeneNest, SpliceNest: Exploring sequence space from genome to protein. Nucleic Acids Res., 30 (1), 299-300. |
| [5] | Schuler,G.D. (1997). Pieces of the puzzle: expressed sequence tags and the catalog of human genes. J. Mol. Med. 75, 694-698. |
| [6] | International Human Genome Sequencing Consortium (2001). Initial sequencing and analysis of the human genome. Nature 409, 860-921. |
| [7] | Kurtz,S., Choudhuri,J.V., Ohlebusch,E., Schleiermacher,C., Stoye,J., and Giegerich,R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633-4642. |
| [8] | Florea,L., Hartzell,G., Zhang,Z., Rubin,G.M., and Miller,W. (1998). A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 8, 967-974. |